光催化氧还原合成H2O2具有安全、清洁和可持续的优点,可利用太阳能驱动反应,减少能量输入与有毒副产物。在众多光催化剂中,二氧化钛(TiO2)因催化活性高、稳定性好、无毒且成本低而备受关注。其成熟的合成工艺也有利于结构调控以提升性能。然而,TiO2在实际应用中仍面临几大挑战:宽带隙导致其只能吸收紫外光,限制了太阳光利用;载流子分离效率低,电子-空穴对易复合,降低量子效率;氧气还原过程中易发生四电子反应生成水,与目标的两电子反应竞争;此外,TiO2表面对H2O2的强吸附和分解作用进一步造成产物损失。 为应对这些问题,研究者采取了多种策略,如负载贵金属纳米颗粒(金、钯)或构建异质结以促进电荷分离;或通过碳材料、聚合物等对TiO₂表面进行钝化,抑制H2O2分解活性位点。尽管这些方法在一定程度上改善了性能,但多数仅解决单一问题,H2O2的整体产率仍不理想。
德国慕尼黑大学Emiliano Cortés教授、中南大学刘敏教授、傅俊伟教授等研究人员设计了Au@TiO2核-多孔壳纳米结构,作为一种综合策略,旨在同时增强H2O2的生成并抑制其分解。Au核嵌入TiO2基体中,在Au-TiO2界面形成肖特基结,有效促进了电荷载流子的分离。更重要的是,Au核的存在引入了局域表面等离子体共振(LSPR),将光吸收扩展到可见光范围,并调控电荷动力学。增强的局部电场由局域表面等离子体共振(LSPR)诱导的热电子产生促进了TiO2中光生电子的定向转移,加速了H2O2的生成。同时, LSPR产生的热电子可以有效地捕获TiO2中的光生空穴,从而抑制H2O2的氧化分解。此外,核壳结构增加了Au和TiO2之间的界面接触面积,进一步促进了界面电荷转移,并增强了催化剂在光催化条件下的结构稳定性。这些协同效应共同促成了高效稳定的光催化H2O2生成。为了更深入地了解该体系,制备了两种Au核形貌的Au@TiO2核壳纳米结构:纳米立方体(NC)和菱形十二面体(RD)。两种材料表现出不同的等离子体响应, NCs比RDs具有更高的电场增强和更高的热载流子生成效率。在3小时的全光谱照射下,两种结构相比裸TiO2均表现出显著的性能提升。NC @TiO2的H2O2产率提高了17倍,生成速率常数kf提高了12倍,分解速率常数kd降低了36% ;而RD@TiO2的产率提高了13倍,生成速率常数kf提高了12倍,分解速率常数kd降低了14%。LSPR 在这些卓越的性能中发挥了关键作用,它能够更有效地利用TiO2中的光生电子-空穴对,促进O2还原为H2O2,同时抑制 H2O2的非预期分解。因此,与单独的紫外光照射相比,额外的LSPR激发使NC@TiO2的H2O2产率提高了52% ,同时kf增大了26%,kd减小了27%;而LSPR响应较弱的 RD@TiO2则表现出适度的增强。
相关研究成果以“Promoting Formation and Suppressing Decomposition of H2O2 via Photocarrier Flow at Au@TiO2 Interfaces”为题发表在J. Am. Chem. Soc.上。

等离子体介导的双向载流子调控机制:通过Au@ TiO2核壳结构中的局域表面等离子体共振,不仅促进光生电子从TiO2向Au的转移以增强H2O2生成,还利用等离子体热电子反向注入TiO2以中和空穴,从而抑制H2O2的分解。
金核形态对等离子体增强效果的依赖性:系统比较了纳米立方与菱形十二面体两种Au核形态,发现NC@ TiO2由于更强的LSPR响应、更高的电场增强和热电子产率,在H2O2产率和稳定性上显著优于RD@ TiO2。
构建了Z-scheme型电荷转移路径:在UV+LSPR双激发条件下,形成类似Z-scheme的电荷流动路径:UV激发的电子从TiO2流向Au促进O2还原,而LSPR热电子从Au流向TiO2消耗空穴,抑制H2O2分解。
系统阐明界面电荷行为:通过XPS、EIS、Mott-Schottky、TRPL、COMSOL模拟、DFT计算等手段,全面揭示了Au与TiO2之间的电子转移方向与程度;LSPR增强电场对电子注入的促进作用;界面态(Au–O–Ti)对空穴的捕获作用。
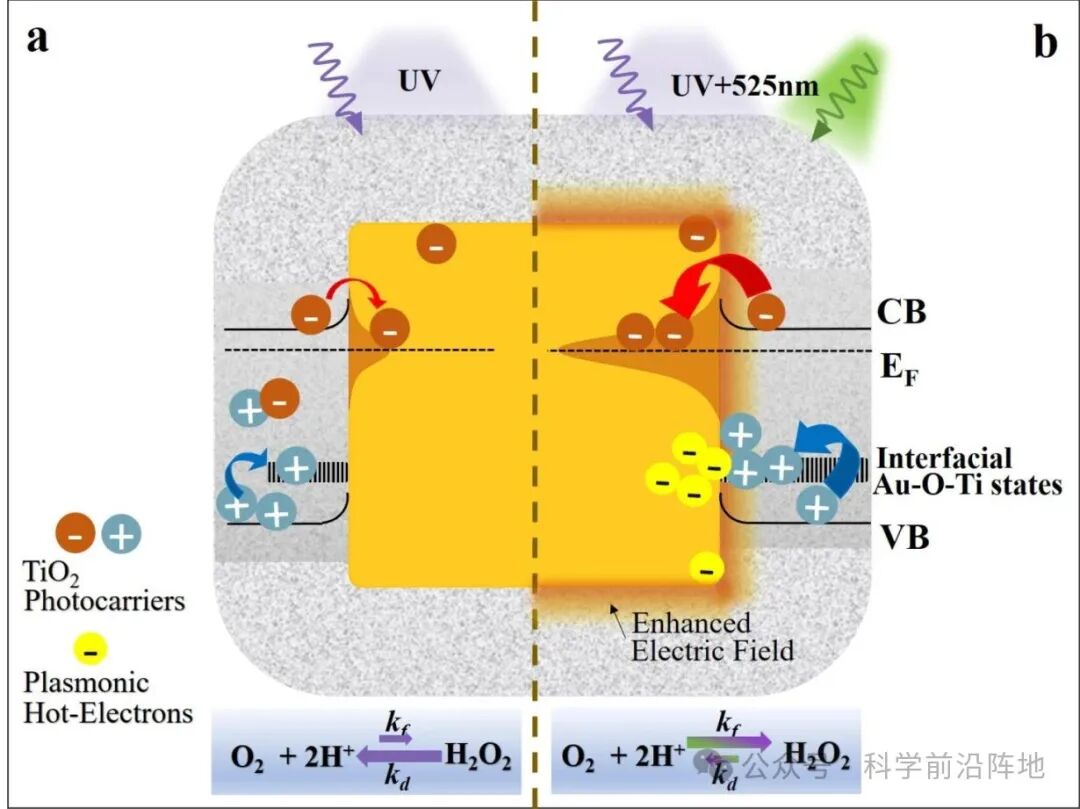
方案1. Au@TiO2杂化纳米体系示意图
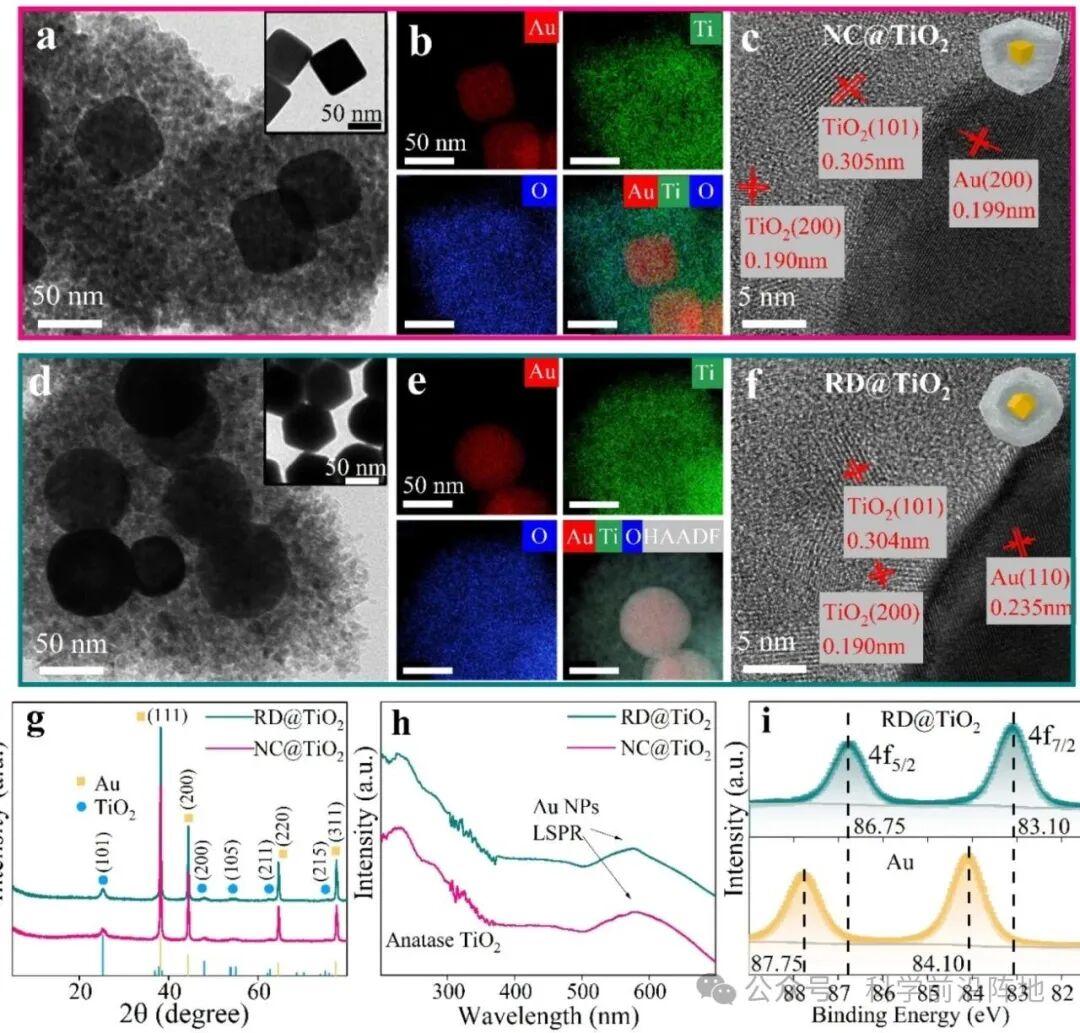
图1. Au@TiO2纳米结构的表征。(a,d) (a) NC@TiO2和(d) RD@TiO2的TEM图像,插图显示了它们各自的Au核。(b,e) (b) NC@TiO2和(e) RD@TiO2的Au、Ti和O元素EDS映射图。(c ,f) (c) NC@TiO2和(f) RD@TiO2的高分辨透射电镜(HRTEM)图谱,红色标记了晶格条纹间距和相应的晶面。(g) NC@TiO2和RD@TiO2的X射线衍射(XRD)图谱,并以面心立方(FCC) Au和锐钛矿相TiO2的参考图谱作为参考(Cu Kα辐射)。(h)NC@ TiO2和RD@TiO2的紫外-可见消光光谱,分别显示出以578 nm和576 nm为中心的特征LSPR吸收峰,以及低于360 nm的TiO2吸收。(i)RD@TiO2和纯Au纳米粒子的Au 4f高分辨率XPS光谱
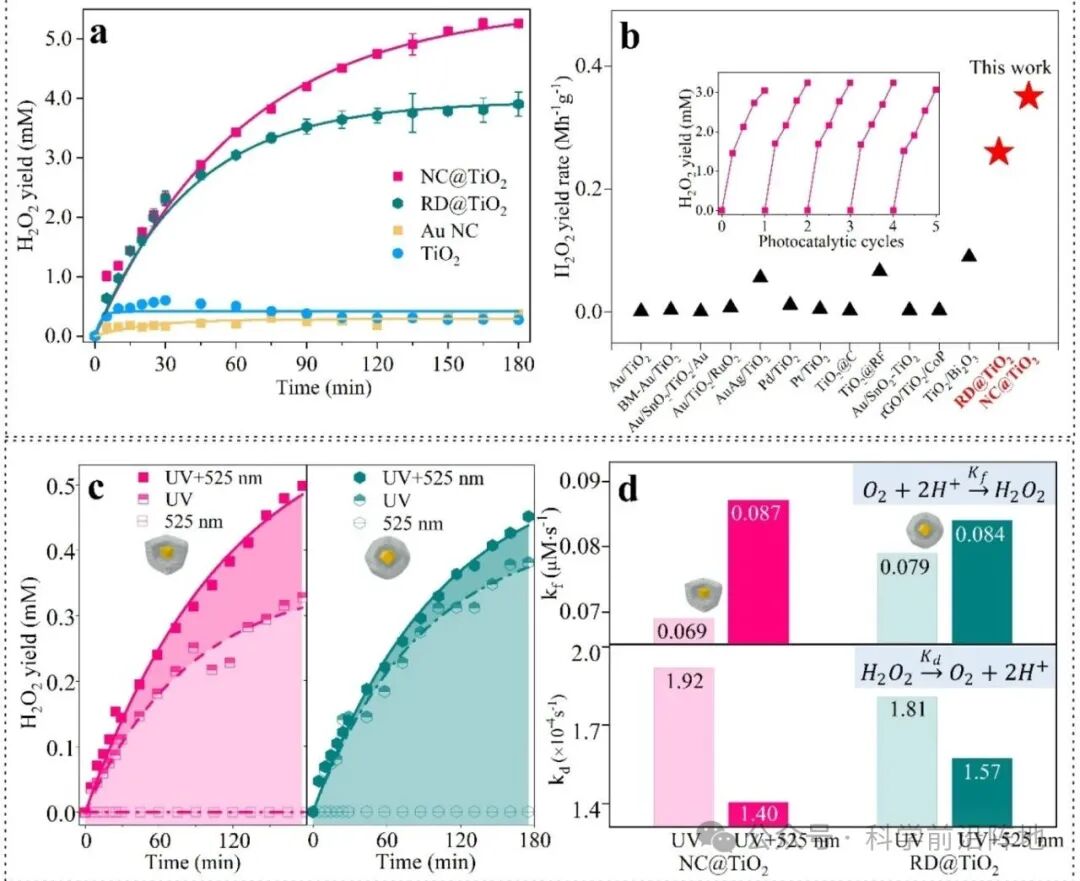
图2. Au@TiO2纳米结构的光催化H2O2生成性能。 (a,b) 氙灯照射下的光催化H2O2生成。(a) NC@TiO2、RD@TiO2、纯Au NPs和锐钛矿型TiO2在3小时内的光催化H2O2生成量随时间的变化。(b)与已报道的光催化剂的H2O2生成速率比较。表S3插图:NC@ TiO2在连续5个1小时循环中的稳定性测试。(c, d)单独紫外光照射和紫外光+525 nm LED联合照射下的光催化H2O2生成。(c) NC@TiO2(左)和RD@TiO2(右)在3小时内的光催化H2O2生成量随时间的变化。(d) H2O2生成速率常数kf(上)和分解速率常数kd(下)
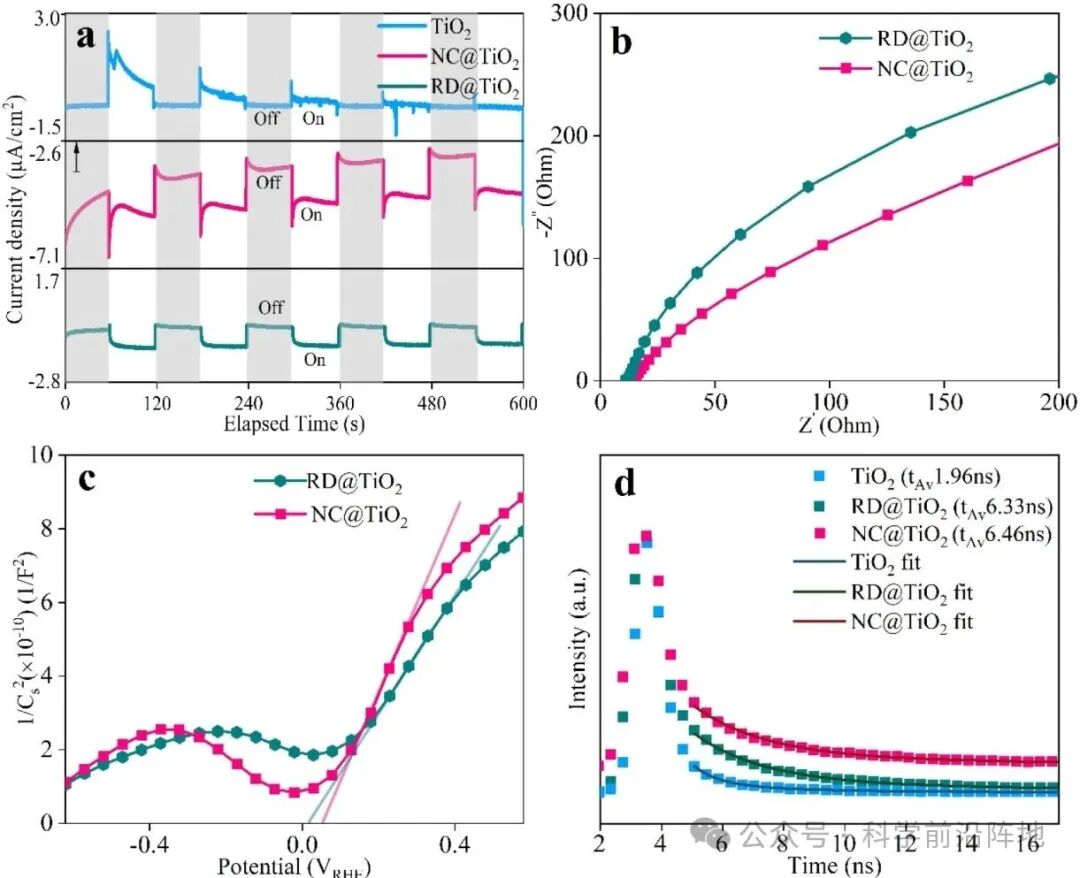
图3. Au@TiO2杂化纳米结构的电子表征。(a) 全光谱照射下的斩波光电流响应,小箭头指示1 μA/cm2。 (b) 光照下的电化学阻抗谱(EIS)奈奎斯特图。(c) 光照下1000 Hz频率下的莫特-肖特基曲线。(d) 时间分辨光致发光(TRPL)衰减曲线
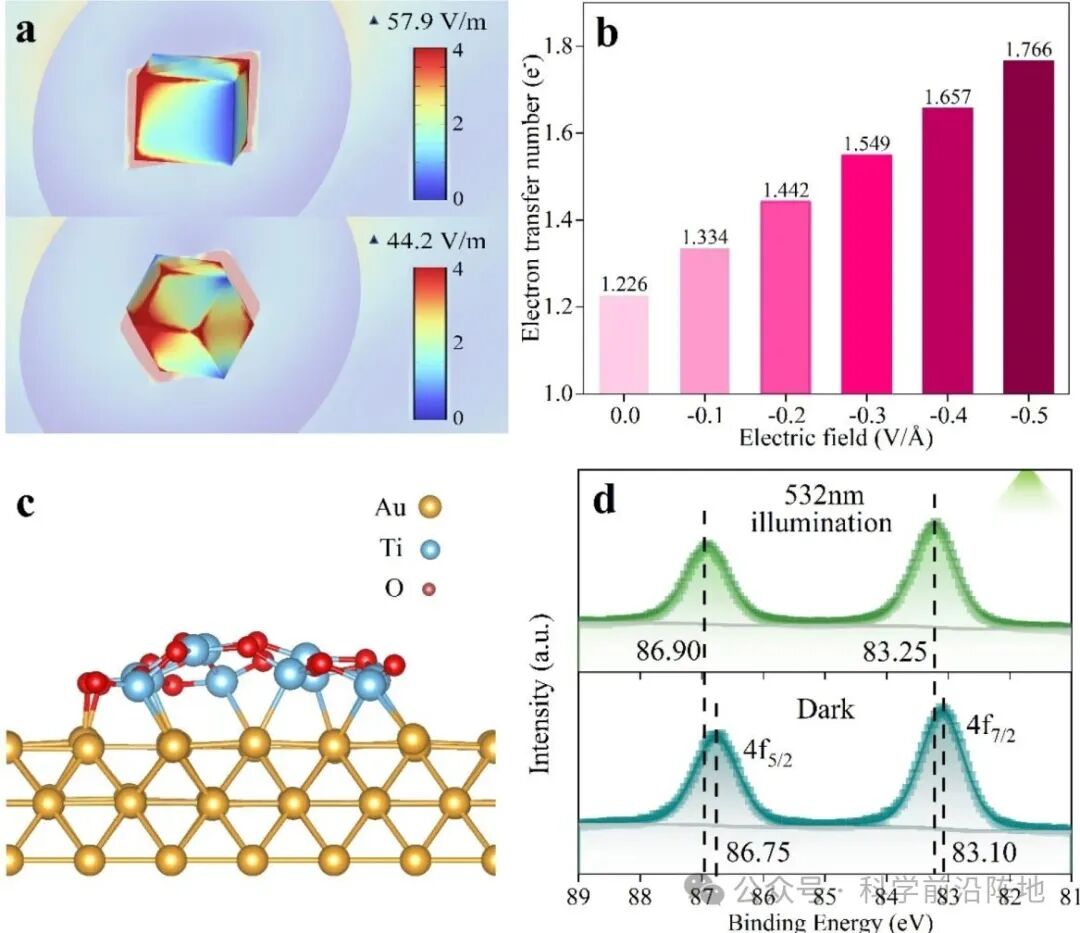
图4.等离子体效应对电子转移过程的影响研究。(a)基于COMSOL Multiphysics有限元分析,模拟等离子体激发下NC@TiO2和RD@TiO2的电场增强。(b) 柱状图展示了电场强度 (0.0–0.5 V/Å) 对电子从TiO2转移到Au的影响,该影响由DFT模拟计算得出。(c) 用于DFT计算的Au@TiO2界面结构模型。(d) RD@TiO2在黑暗条件和532 nm光照下Au 4f的高分辨率XPS光谱
这项研究表明,等离子体Au@TiO2核壳纳米结构通过主动调控光载流子动力学,克服了光催化合成H2O2的双重挑战——生成效率低和分解速度快。Au核的局域表面等离子体共振(LSPR)激发重塑了金属-半导体界面处的电荷流动:TiO2中的光生电子被引导至Au位点以驱动O2还原,同时等离子体热电子捕获TiO2中的空穴,抑制氧化性H2O2分解。这种双重效应表明,LSPR并非被动的光捕获现象,而是控制反应选择性的关键因素。形貌依赖性研究进一步表明,具有更强等离子体场的纳米立方体金核比菱形十二面体金核表现出更优异的活性。这种颗粒几何形状、等离子体强度和催化效果之间的关联凸显了纳米尺度光学设计在决定催化行为方面的核心作用,而这种见解在传统光催化剂中却鲜有涉及。除了为基于TiO2的光催化H2O2生成设定新的基准速率外,这些发现还建立了一个可推广的设计原则:等离子体纳米结构可以被定制,从而引导电荷载流子沿着所需的路径运动,进而将太阳能到化学能的转化效率与选择性相结合。这一概念上的突破使等离子体光催化不仅成为可持续H2O2生成的一个极具前景的平台,而且在人工光合作用和绿色化学制造等领域也具有更广泛的应用前景。
原文链接:
https://pubs.acs.org/doi/10.1021/jacs.5c14650
声明:仅代表作者个人观点,作者水平有限,如有不科学之处,请在下方留言指正!

来源|科学前沿阵地
